# %pip install openmm
# %pip install matplotlib
from openmm import app, unit, Vec3
import openmm
import matplotlib.patches as mpatches
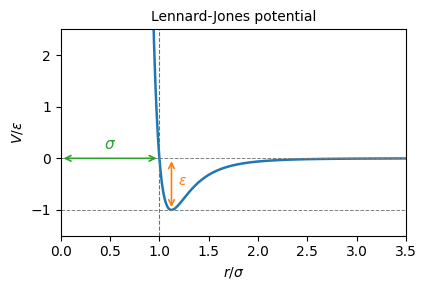
def simulate_water_dimer(
d0: float = 1,
v_approach: float = 0.04,
n_steps: int = 7500,
subsample: int = 50,
) -> tuple[np.ndarray, np.ndarray, np.ndarray]:
"""TIP3P water dimer via OpenMM Verlet.
Parameters
----------
d0 Initial O-O distance (nm).
v_approach Initial approach speed of each molecule (nm/ps).
n_steps Total integration steps (1 fs each).
subsample Record every `subsample` steps.
Returns
-------
pos_traj shape (T, 6, 3) in Angstrom
ke_traj shape (T,) in kJ/mol
pe_traj shape (T,) in kJ/mol
"""
# topology: 2 water molecules, each with 1 O and 2 H atoms, and bonds between them
top = app.Topology()
chain = top.addChain()
for _ in range(2):
# for each water molecule (2 in total)
res = top.addResidue('HOH', chain)
O = top.addAtom('O', app.Element.getBySymbol('O'), res)
H1 = top.addAtom('H1', app.Element.getBySymbol('H'), res)
H2 = top.addAtom('H2', app.Element.getBySymbol('H'), res)
top.addBond(O, H1)
top.addBond(O, H2)
# use the TIP3P water model for forces and energies
ff = app.ForceField('tip3p.xml')
system = ff.createSystem(
top,
nonbondedMethod=app.NoCutoff,
constraints=app.HAngles,
)
# Remove center-of-mass motion remover (not desired for small systems)
for i in reversed(range(system.getNumForces())):
if isinstance(system.getForce(i), openmm.CMMotionRemover):
system.removeForce(i)
# water geometry parameters
r_OH = 0.09572 # O-H bond length (nm)
a = 104.52 * np.pi / 180 # H-O-H angle (radians)
c52 = np.cos(a / 2) # cosine of half the angle
s52 = np.sin(a / 2) # sine of half the angle
# initial positions of the 6 atoms (2 waters x 3 atoms each)
# "H" of molecule 0 points towards "O" of molecule 1
# all positions with z = 0
positions = unit.Quantity([
Vec3(0.0, 0.0, 0),
Vec3(r_OH, 0.0, 0),
Vec3(-r_OH * c52, r_OH * s52, 0),
Vec3(d0, 0.0, 0),
Vec3(d0 + r_OH * c52, r_OH * s52, 0),
Vec3(d0 + r_OH * c52, -r_OH * s52, 0),
], unit.nanometer)
integrator = openmm.VerletIntegrator(1.0 * unit.femtosecond)
sim = app.Simulation(top, system, integrator)
sim.context.setPositions(positions)
# initial velocities: both molecules approach each other along x-axis
sim.context.setVelocities(unit.Quantity([
Vec3( v_approach, 0, 0), Vec3( v_approach, 0, 0), Vec3( v_approach, 0, 0),
Vec3(-v_approach, 0, 0), Vec3(-v_approach, 0, 0), Vec3(-v_approach, 0, 0),
], unit.nanometer / unit.picosecond))
# frames to save (after subsampling)
T = n_steps // subsample
# allocate arrays for trajectory
pos_traj = np.zeros((T, 6, 3))
ke_traj = np.zeros(T)
pe_traj = np.zeros(T)
# integration loop
for step in range(n_steps):
# make one integration step
sim.step(1)
# save trajectory at subsampled steps
if step % subsample == 0:
idx = step // subsample
if idx < T:
s = sim.context.getState(getPositions=True, getEnergy=True)
pos_traj[idx] = s.getPositions(asNumpy=True).value_in_unit(unit.angstrom)
ke_traj[idx] = s.getKineticEnergy().value_in_unit(unit.kilojoule_per_mole)
pe_traj[idx] = s.getPotentialEnergy().value_in_unit(unit.kilojoule_per_mole)
return pos_traj, ke_traj, pe_traj
def make_water_animation(
pos_traj: np.ndarray,
ke_traj: np.ndarray,
pe_traj: np.ndarray,
dt_frame: float, # in ps
interval: int = 50, # in ms between frames in the animation
) -> animation.FuncAnimation:
# time points for plotting energy
t = np.arange(len(pos_traj)) * dt_frame
# total energy (total = ke + pe)
total = ke_traj + pe_traj
# two axes:
# - left: simulation frame
# - right: energies over time
fig, (ax_m, ax_e) = plt.subplots(
1, 2, figsize=(5.5, 3), gridspec_kw={'width_ratios': [1.2, 1]}
)
# simulation frame
ax_m.set_aspect('equal')
ax_m.set_xlim(-1.5, 11.5)
ax_m.set_ylim(-3.0, 3.0)
ax_m.set_axis_off()
ax_m.set_title('TIP3P water dimer', fontsize=9)
r_O, r_H = 0.66, 0.31 # display radii in Å
o_colors = ['C3', 'steelblue']
# atom patches — created once, positions updated each frame
o_patches = [
ax_m.add_patch(mpatches.Circle(
pos_traj[0, 3 * mol, :2], r_O,
lw=1.2, edgecolor='black', facecolor=o_colors[mol], zorder=3
))
for mol in range(2)
]
h_patches = [
ax_m.add_patch(mpatches.Circle(
pos_traj[0, 3 * mol + hi, :2], r_H,
lw=0.8, edgecolor='gray', facecolor='white', zorder=3
))
for mol in range(2) for hi in [1, 2]
]
bond_lines = [ax_m.plot([], [], 'k-', lw=1.5, zorder=2)[0] for _ in range(4)]
hbond_line = ax_m.plot([], [], 'b--', lw=1.0, alpha=0.8, zorder=1)[0]
# energy plot
ax_e.set_xlim(0, t[-1])
ax_e.set_ylim(min(pe_traj.min(), -25), max(ke_traj.max(), 1) * 1.3)
ax_e.set_xlabel('time (ps)', fontsize=8)
ax_e.set_ylabel('energy (kJ/mol)', fontsize=8)
ax_e.tick_params(labelsize=7)
ax_e.axhline(total[0], color='gray', lw=0.7, ls=':')
ke_line = ax_e.plot([], [], color='C1', lw=1, label='kinetic energy')[0]
pe_line = ax_e.plot([], [], color='C4', lw=1, label='potential energy')[0]
tot_line = ax_e.plot([], [], color='black', lw=1.2, ls='--', label='total energy')[0]
ax_e.legend(fontsize=6, loc='upper right')
plt.tight_layout()
# scale bar: 1 nm = 10 Å
_sb = 10.0
_sx, _sy = -1.2, -2.7
ax_m.plot([_sx, _sx + _sb], [_sy, _sy], 'k-', lw=2, solid_capstyle='butt')
ax_m.plot([_sx, _sx], [_sy - 0.1, _sy + 0.1], 'k-', lw=1.5)
ax_m.plot([_sx + _sb, _sx + _sb], [_sy - 0.1, _sy + 0.1], 'k-', lw=1.5)
ax_m.text(_sx + _sb / 2, _sy - 0.15, '1 nm', ha='center', va='top', fontsize=7)
# animation callback for frame update
def update(frame: int) -> list:
# update atom positions
for mol in range(2):
base = 3 * mol
o_patches[mol].set_center(pos_traj[frame, base, :2])
h_patches[2 * mol ].set_center(pos_traj[frame, base + 1, :2])
h_patches[2 * mol + 1].set_center(pos_traj[frame, base + 2, :2])
for hi, bl in enumerate(bond_lines[2 * mol: 2 * mol + 2]):
xy = np.array([pos_traj[frame, base, :2],
pos_traj[frame, base + 1 + hi, :2]])
bl.set_data(xy[:, 0], xy[:, 1])
# H-bond indicator:
# closest H of mol 0 to O of mol 1
h_dists = [np.linalg.norm(pos_traj[frame, h] - pos_traj[frame, 3])
for h in [1, 2]]
best_h = 1 + int(np.argmin(h_dists))
if min(h_dists) < 2.8: # 2.8 Å H-bond cutoff
xy = np.array([pos_traj[frame, best_h, :2], pos_traj[frame, 3, :2]])
hbond_line.set_data(xy[:, 0], xy[:, 1])
else:
hbond_line.set_data([], [])
s = slice(None, frame + 1)
ke_line.set_data(t[s], ke_traj[s])
pe_line.set_data(t[s], pe_traj[s])
tot_line.set_data(t[s], total[s])
return o_patches + h_patches + bond_lines + [hbond_line, ke_line, pe_line, tot_line]
anim = animation.FuncAnimation(fig, update, frames=len(pos_traj), interval=interval)
plt.close(fig)
return anim